![]()
Overview
The biodiversity patterns that we currently observe on Earth have been continuously shaped by evolutionary dynamics over longer periods of time. Thus, the historical dynamics experienced by a particular clade during its evolution are crucial to understand its current patterns, and many of these significant historical influences are widely distributed and imprinted across extant phylogenies. Alternatively, solely focusing on macroevolution dynamics overlooks important and complex patterns that emerge due to biogeographical processes. For instance, these dynamics may differ among assemblages due to various constraints experienced over time, resulting in distinct phylogenetic histories stored within them. However, dissecting phylogenies to comprehend the contributions of cladogenesis events from different depths on a given pattern has been challenging until now, given the lack of tools readily providing this type of information.
The primary goal of treesliceR is to provide a wide range of functions capable of cutting phylogenies at any evolutionary depth (Figure 1). These cutted phylogenies can be used by users to explore various evolutionary patterns stored at different temporal depths, as well as any other information derived from these pruned phylogenies. To ensure a more accessible and user-friendly processes for obtaining these pruned phylogenies, treesliceR functions offer multiple criteria for making slices based on users requirements. For instance, it enables users to cut phylogenies in different orientations, such as ‘rootwardly’ (from root to tips) and ‘tipwardly’ (from tips to its root), or simply obtain a phylogenetic slice within a specific time interval of interest. Alternatively, multiple phylogenetic slices of equal width can be produced, similar to a time-series, based on user-input temporal criteria, which can be expressed in either millions of years or in terms of phylogenetic diversity.
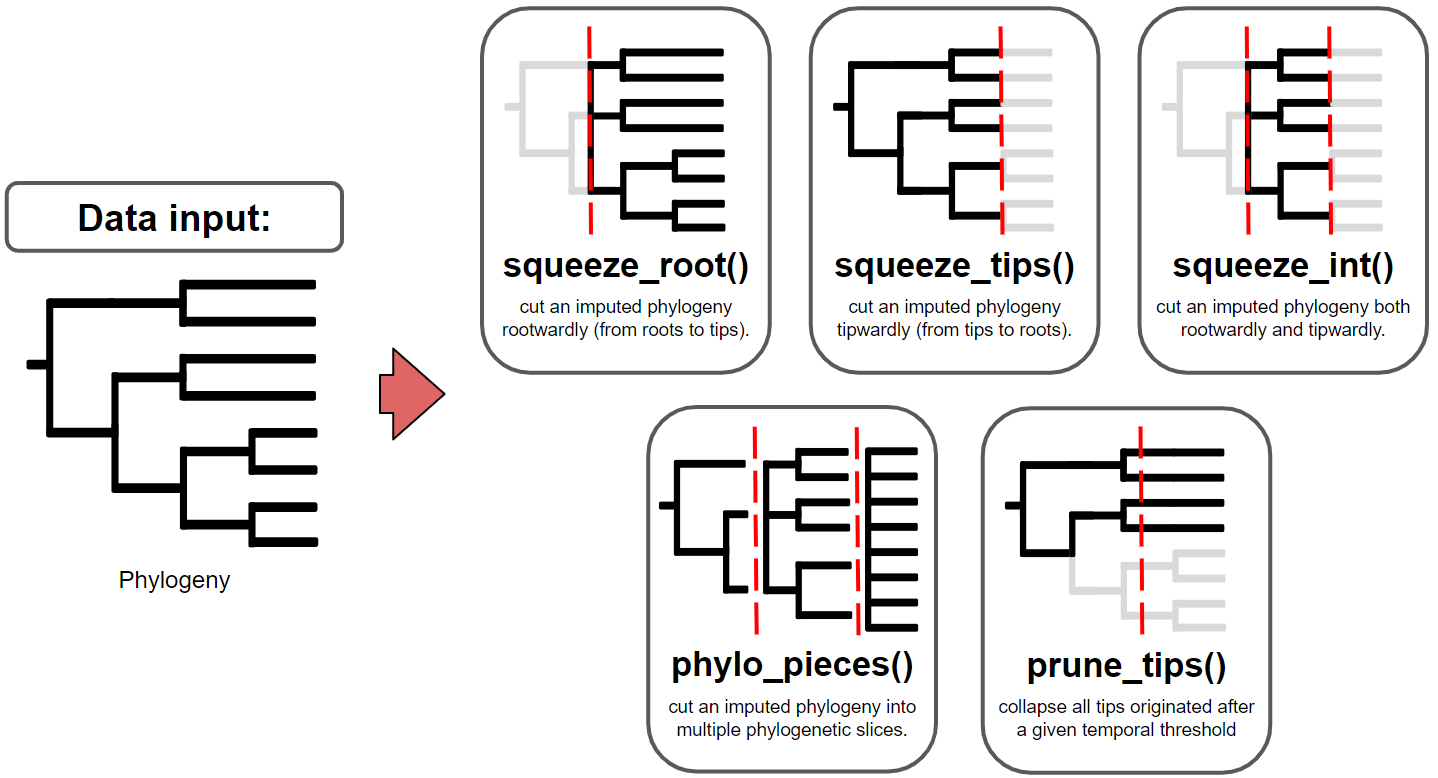
Figure 1: This figure provides illustrated examples of the functions for slicing phylogenies available within treesliceR. The red dashed lines indicate hypothetical temporal thresholds input by the user.
The second goal of treesliceR is to provide functions that facilitate the assessment of a novel family of rates of accumulation through time for any phylogenetic index (Figure 2). The rationale behind these rates is that most currently available indexes do not differentiate the contributions of lineages at different depths to a given observed pattern. This implies that the same value of a particular phylogenetic index can be obtained through different configurations of species compositions and tree topologies. For instance, a high phylogenetic diversity observed within an assemblage could result from a few deep lineages or a high richness of recent lineages. Therefore, treesliceR offers functions for calculating the accumulation of a given phylogenetic pattern (e.g., PD, PE, Pβ…) through time to elucidate the main lineages depths responsible for generating it. For more detailed information regarding the mathematical procedures behind these rates, please refer to Araujo et al. (2025).
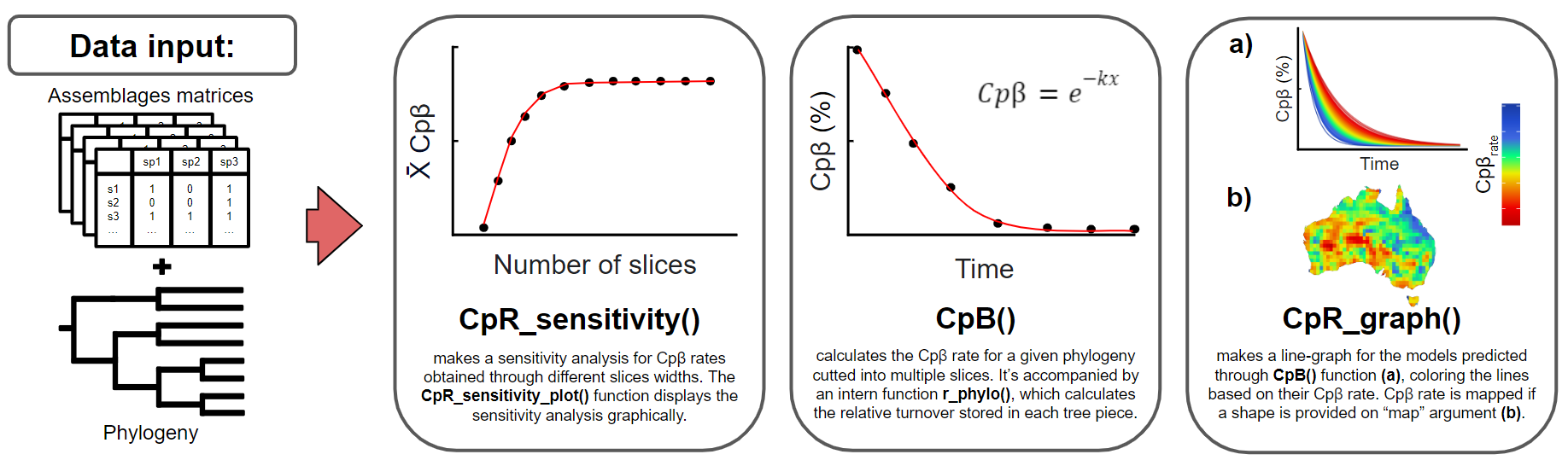
Figure 2: This figure provide illustrated examples of the available functions for calculating rates of accumulation of phylogenetic indexes, assessing their most parsimonious number of slices to model these rates (i.e., sensitivity analysis), and presenting their graphical outputs. The figure demonstrates the functions for obtaining only the rates of accumulations of phygeloenetic β-diversity (Cpβ) as an example, but these rates can also be applied to other phylogenetic indexes (for more details, see treesliceR CRAN).
treesliceR is an easy-to-use and open-access R package that is continuously evolving, with the goal of becoming a standard toolbox for users interested in temporally dissecting any phylogenetic information. It strives to continually develop innovative methods to reveal previously concealed phylogenetic patterns over the evolutionary time. The package was developed following user-friendly software development practices, offering functions with core parallelization, detailed error messages in case of failures, and graphical functions that can rapidly generate ready-to-publish figures. Nevertheless, treesliceR is freely maintained on GitHub, where its user community can directly engage with maintainers to report bugs, contribute improvements to our existing algorithms through pull request, or even propose novel ideas for future functions.
Installation
If you are interested in using the treesliceR tools in your classes or research, you can install the CRAN (Comprehensive R Archive Network) version simply by typing:
install.packages("treesliceR")Alternatively, you can install the development version of treesliceR from GitHub by typing:
devtools::install_github("AraujoMat/treesliceR")Now, you are ready to temporally dissect any phylogenetic pattern of interest! For more detailed information, visit our CRAN!
Examples
To learn about various ways to use and solve problems using treesliceR, you can explore some of our vignettes:
- Introduction to treesliceR: This section provides straightforward and beginner-friendly applications with simple illustrations of the tools available within the package;
- Passerines distribution: Here, you’ll find an applied example of our tools focusing on Australian passeriformes, where we compare species richness distributions between older and more recent lineages;
- Passerines diversification: In this vignette, we replicate the case study by Araujo et al. (2025). It presents the workflow executed for both a single phylogeny and multiple sampled phylogenies for passeriformes.
Getting help
Encountering issues with certain treesliceR functions on your computer? Please report these bugs with a minimal reproducible example on the package’s GitHub page. If you have suggestions for improving our algorithms, don’t hesitate to submit your ideas through pull requests.
If you have intriguing ideas for exploring phylogenetic information temporally, please feel free to contact us via email at: matheusaraujolima@live.com. You can track our commit updates and version controls directly on GitHub.