Prelude
treesliceR is an R package that offers versatile tools
for subsetting and slicing phylogenetic trees at various depths and
orientations. Sliced and pruned phylogenies generated with
treesliceR are readily available for downstream analysis,
facilitating the evaluation of phylogenetic patterns such as alpha and
beta phylogenetic metrics. With treesliceR, phylogenies can
be sliced in different temporal orientations, either “rootwardly” or
“tipwardly”, and internally using diverse temporal criteria such as
million od years, accumulated phylogenetic diversity, or quantiles of
tip ages distribution.
Moreover, treesliceR contains functions for assessing
the rates of accumulation of phylogenetic information (e.g., “α” and “β”
diversities) over time. For a comprehensive introduction to calculating
these metrics, refer to this
vignette. All analysis are designed to be flexible, reliable, and
efficient, with computationally time-demanding functions optimized for
parallel computation. Additionally, our package includes plotting
functions that enable the production of ready-to-use figures.
To demonstrate the basic usage of treesliceR, we will
provide examples illustrating how to subset and slice phylogenies in
different ways. To begin, it is necessary to load the ape
package:
Now, we’ll need to load (and install if necessary) the
treesliceR package:
# Loading it
library(treesliceR)To showcase the flexibility of treesliceR, we will
provide a demonstration on how to subset and slice a phylogenetic tree
for passarine species (308 species) included in the package (obtained
from Jetz et
al. (2012)). The pass_tree object comprises a
multiphylo object containing 100 passerine trees. For simplicity, we
will focus on a single tree. To load this dataset, we use the following
code:
tree <- pass_trees[[1]]1. Pruning function
treesliceR encompasses functions for pruning a
phylogenetic tree based on a specific absolute tree depth or by
establishing a quantile value devied from the age distribution of the
last splitting events for the tips of a given phylogenetic tree. In both
procedures (absolute age or quantile), all tips with the age of the last
splitting event after or before the
value specified in the time argument will be pruned or retained in the
phylogeny (depending on the method argument). In the next
examples, we will illustrate how to prune the phylogenetic tree using an
absolute depth (in this case, in million of years) or a scalar
indicating a quantile drawn from the tip-species age distribution.
1.1. Pruning using absolute depth
First, let’s prune the phylogeny to retain only species with splitting events older than 10 and 30 millions years:
tree_pruned10 <- prune_tips(tree = tree, time = 10, qtl = F) # keep species older than 10my
tree_pruned30 <- prune_tips(tree = tree, time = 30, qtl = F) # keep species older than 30myLets check the format of those phylogenetic trees:
oldpar <- par(mfrow = c(1, 3)) # Setting an 1x3 graphical display
plot(tree, main = "All species", show.tip.label = F); axisPhylo()
plot(tree_pruned10, main = "Species older than 10my", show.tip.label = F); axisPhylo()
plot(tree_pruned30, main = "Species older than 30my", show.tip.label = T); axisPhylo()
par(oldpar) # Returning to the original display
We can see that only tip-species with the last splitting events older than 10 and 30 million years were retained in the trees. It is important to note that the original branch lengths remain unchanged. From left to right, we have the original phylogeny, the phylogeny containing only species with the last splitting event older than 10 million years, and finally, the phylogeny with species where the last branching event is older than 30 million years (only three species in our example).
Conversely, the inverse procedure can also be done, allowing us to
keep only the species with the last branching event younger than the
time argument by setting the method argument
as 2. Therefore:
tree_pruned10_inverse <- prune_tips(tree = tree, time = 10, qtl = F, method = 2) # keep species younger than 10my
tree_pruned30_inverse <- prune_tips(tree = tree, time = 30, qtl = F, method = 2) # keep species younger than 30my
# plotting phylogenies
oldpar <- par(mfrow = c(1, 3)) # Setting an 1x3 graphical display
plot(tree, main = "All species", show.tip.label = F); axisPhylo()
plot(tree_pruned10_inverse, main = "Species younger than 10my", show.tip.label = F); axisPhylo()
plot(tree_pruned30_inverse, main = "Species younger than 30my", show.tip.label = F); axisPhylo()
par(oldpar) # Returning to the original display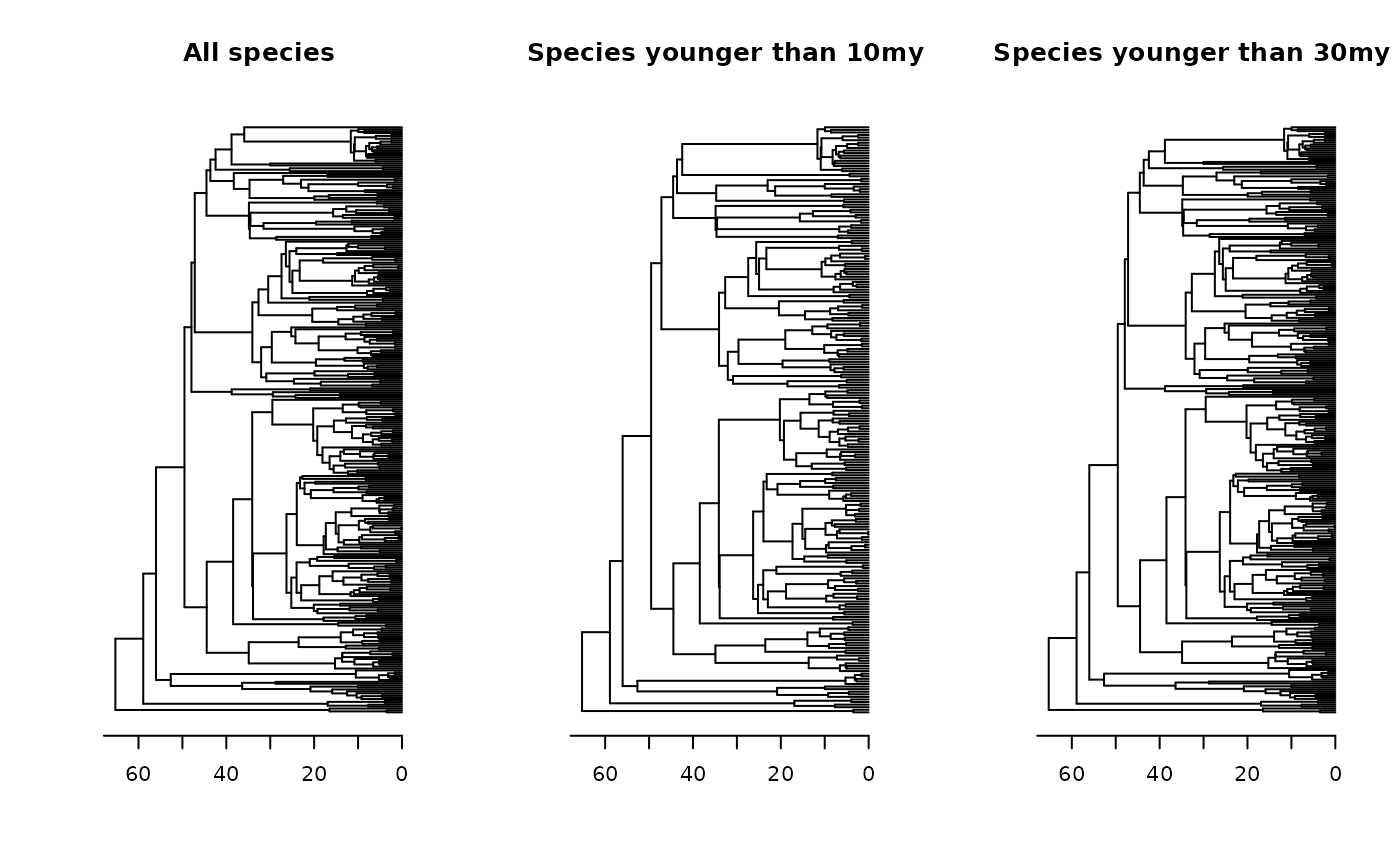
1.2. Pruning using depth quantiles
The same procedure can be applied by setting the time
argument as a scalar indicating the quantile of the distribution of
tip-species ages, given by the time of the last branching event. For
instance, to retain only species with age values younger (lower) than
the ages of 25th quantile of the age distribution of all species, we can
do the following:
tree_pruned25q <- prune_tips(tree, 0.25, qtl = T, method = 2)
plot(tree_pruned25q, main = "Species with ages younger than 25th quantile", show.tip.label = F); axisPhylo()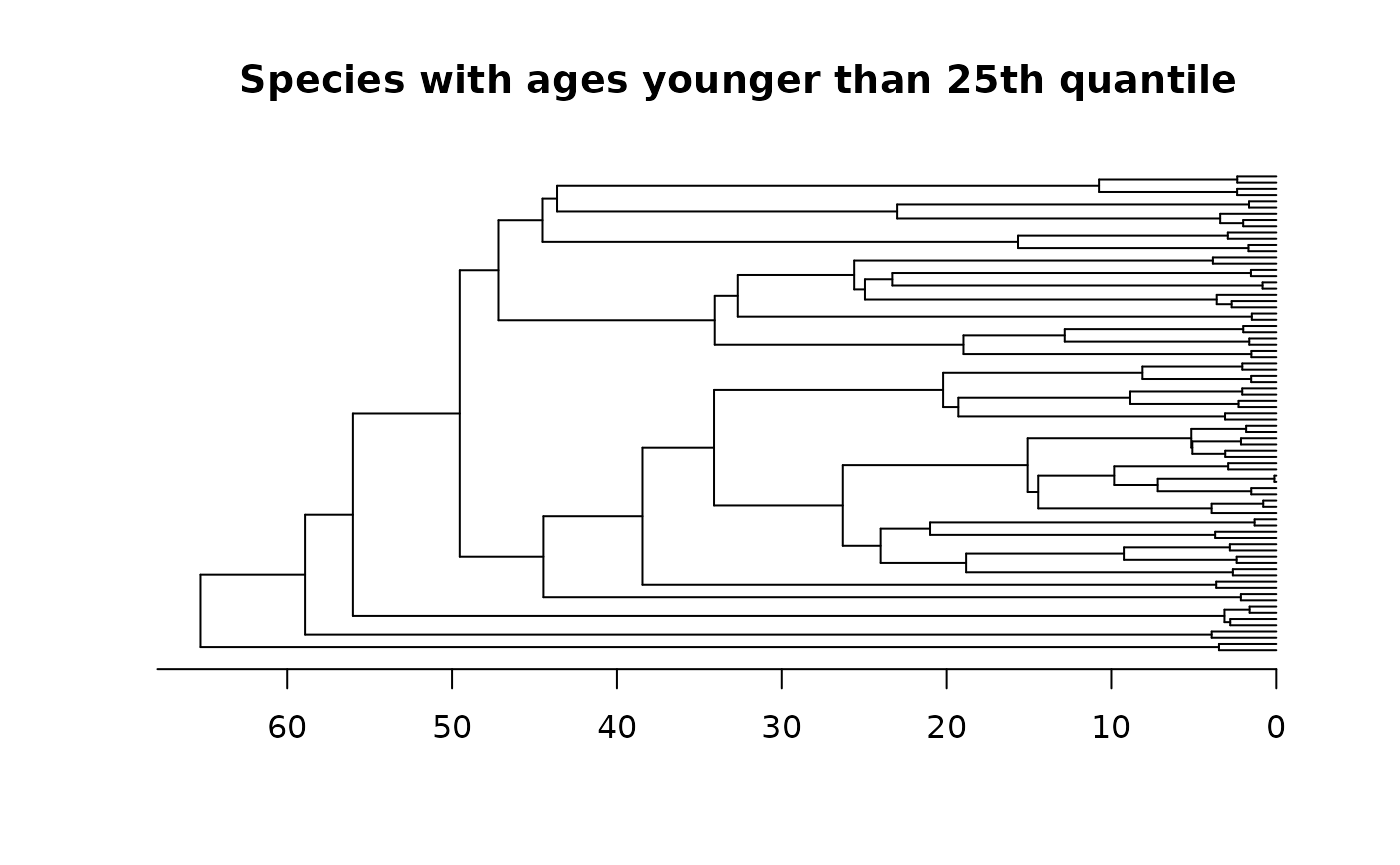
Note that if we aim to retain species with ages younger than the 25th
quantile, the method argument must be set as 2, as
illustrated in the above example. The default value is 1, signifying the
removal of tips with ages younger than the threshold specified in the
time argument.
2. Slicing functions
The second family of functions present within treesliceR
enables the slicing of phylogeny in vairous ways. In this section, we
will show the flexibility of treesliceR in performing tree
slicing and visualizing the output of these functions.
2.1. Slicing from the tips to the root
The first option to slice phylogenies is by squeezing its branch
lengths from the tips to the root using the function
squeeze_tips(). Squeezing, in this context, refers to
collapsing branch lengths based on a given threshold (tree depth, in
this case, in million years). This function requires providing a
phylogenetic tree and a numeric value indicating the depth at which the
branches will be compressed. For instance, let’s squeeze the passerine
phylogeny at three different ages (10, 30 and 50 million years):
tree_squeeze10 <- squeeze_tips(tree = tree, time = 10)
tree_squeeze30 <- squeeze_tips(tree = tree, time = 30)
tree_squeeze50 <- squeeze_tips(tree = tree, time = 50)We can visualize the phylogeny before and after the squeezing process to check out the differences:
oldpar <- par(mfrow = c(1, 3)) # Setting an 1x3 graphical display
plot(tree_squeeze10, main = "squeezed at 10my", show.tip.label = F); axisPhylo()
plot(tree_squeeze30, main = "Squeezed at 30my", show.tip.label = F); axisPhylo()
plot(tree_squeeze50, main = "Squeezed at 50my", show.tip.label = F); axisPhylo()
par(oldpar) # Returning to the original display
It is noteworthy that setting the dropNodes argument as
TRUE (default is FALSE) alters the tree structure by removing nodes with
no branch lengths from the original tree. We can check this out by
typing:
tree_squeeze30_drop <- squeeze_tips(tree = tree, time = 30, criterion = "my", dropNodes = TRUE)
tree_squeeze30 # full binary tree
#>
#> Phylogenetic tree with 308 tips and 307 internal nodes.
#>
#> Tip labels:
#> Pitta_iris, Pitta_versicolor, Pitta_erythrogaster, Atrichornis_clamosus, Atrichornis_rufescens, Menura_alberti, ...
#>
#> Rooted; includes branch length(s).
tree_squeeze30_drop # tree with nodes dropped
#>
#> Phylogenetic tree with 308 tips and 58 internal nodes.
#>
#> Tip labels:
#> Pitta_iris, Pitta_versicolor, Pitta_erythrogaster, Atrichornis_clamosus, Atrichornis_rufescens, Menura_alberti, ...
#>
#> Rooted; includes branch length(s).You’ll observe that the tree_squeeze30 object forms a
full binary tree (308 species and 307 nodes), while the object
tree_squeeze30_drop is not a binary tree, as nodes younger
than 30 million years were dropped out from the tree.
As mentioned before, the tree can be squeezed using different depths criteria. Let’s explore this by using accumulated phylogenetic diversity (PD) over the years to squeeze and plotting all trees to observe the differences. First, lets assess the total PD available within the tree:
PD_total <- sum(tree$edge.length)Then, lets squeeze our tips to the point where 10% and 50% of the total PD was accumulated in the phylogenetic tree:
tree_squeeze10 <- squeeze_tips(tree = tree, time = PD_total/10, criterion = "pd")
tree_squeeze50 <- squeeze_tips(tree = tree, time = PD_total/2, criterion = "pd")
oldpar <- par(mfrow = c(1, 2)) # Setting an 1x2 graphical display
plot(tree_squeeze10, main = "Tree squeezed at 10% PD", show.tip.label = F); axisPhylo()
plot(tree_squeeze50, main = "Tree squeezed at 50% PD", show.tip.label = F); axisPhylo()
par(oldpar) # Returning to the original display
2.2. Slicing from the root to the tips
Another option available in treesliceR is the
squeeze_root() function, allowing users to squeeze the
phylogenetic tree from the root to the tips using the same depth
criterion and arguments as demonstrated in the previous section. For
instance, let’s squeeze our passerine phylogeny root to 50 million
years:
tree_root50 <- squeeze_root(tree = tree, time = 50)
plot(tree_root50, main = "Tree sliced rootwardly in 50my", show.tip.label = F); axisPhylo()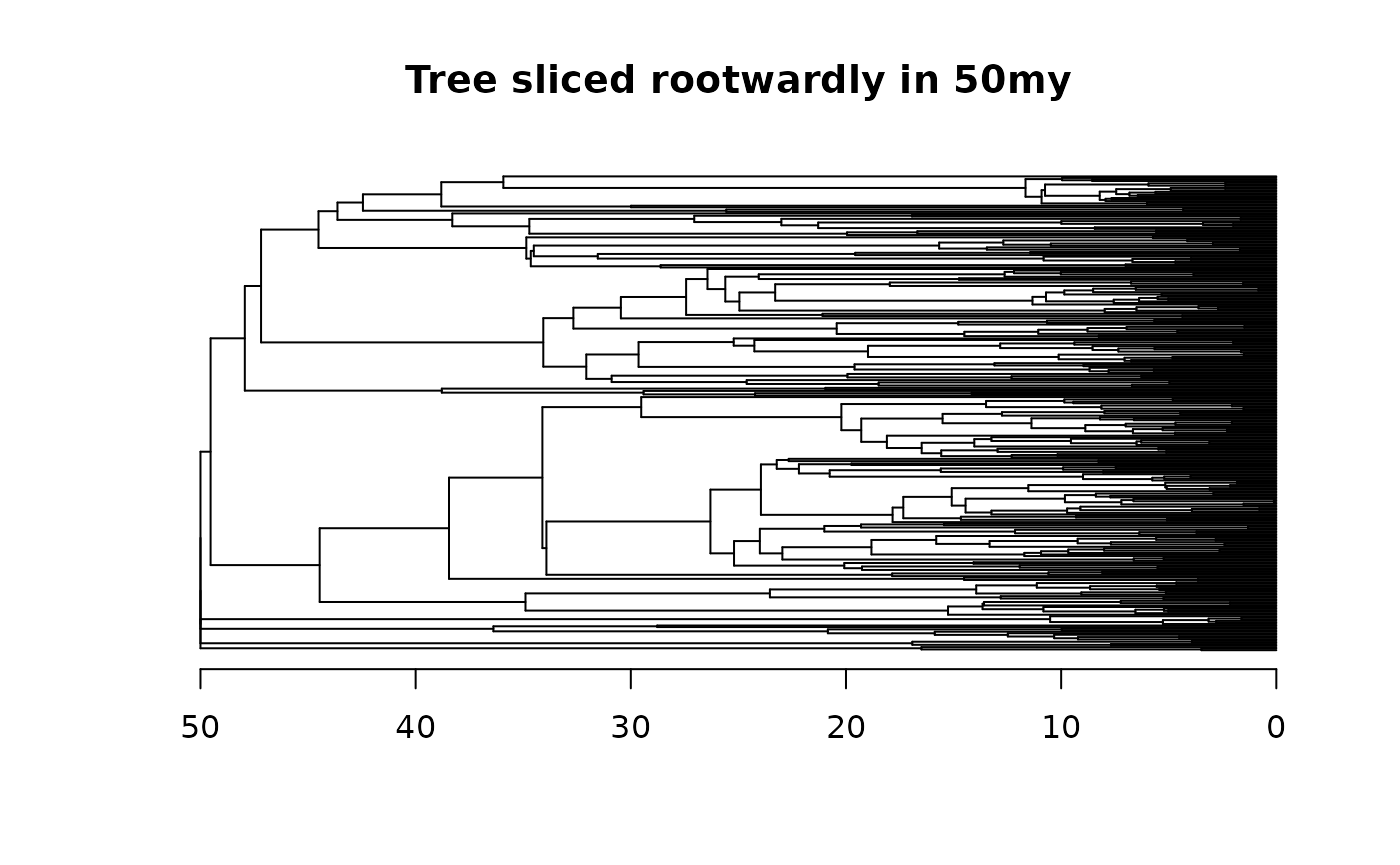
Notice that the root of the tree now begins at 50 millions of years.
2.3. Slicing phylogenies internally
treesliceR also provides the option to slice phylogenies
internally by using the function squeeze_int(). In this
function, the user must specify two values representing the depth
boundaries of the tree that will be retained (or removed) from the
phylogeny.
For example, if we want to keep only branches of the tree lying between 30 and 10 million years, we can use the following code:
tree_int <- squeeze_int(tree = tree, from = 30, to = 10,)Let’s check out the slice of the tree that was retained and compare it with the original tree:
oldpar <- par(mfrow = c(1, 2)) # Setting an 1x2 graphical display
plot(tree, main = "Original tree", show.tip.label = F); axisPhylo()
plot(tree_int, main = "Tree slice 10-30my", show.tip.label = F); axisPhylo()
par(oldpar) # Returning to the original display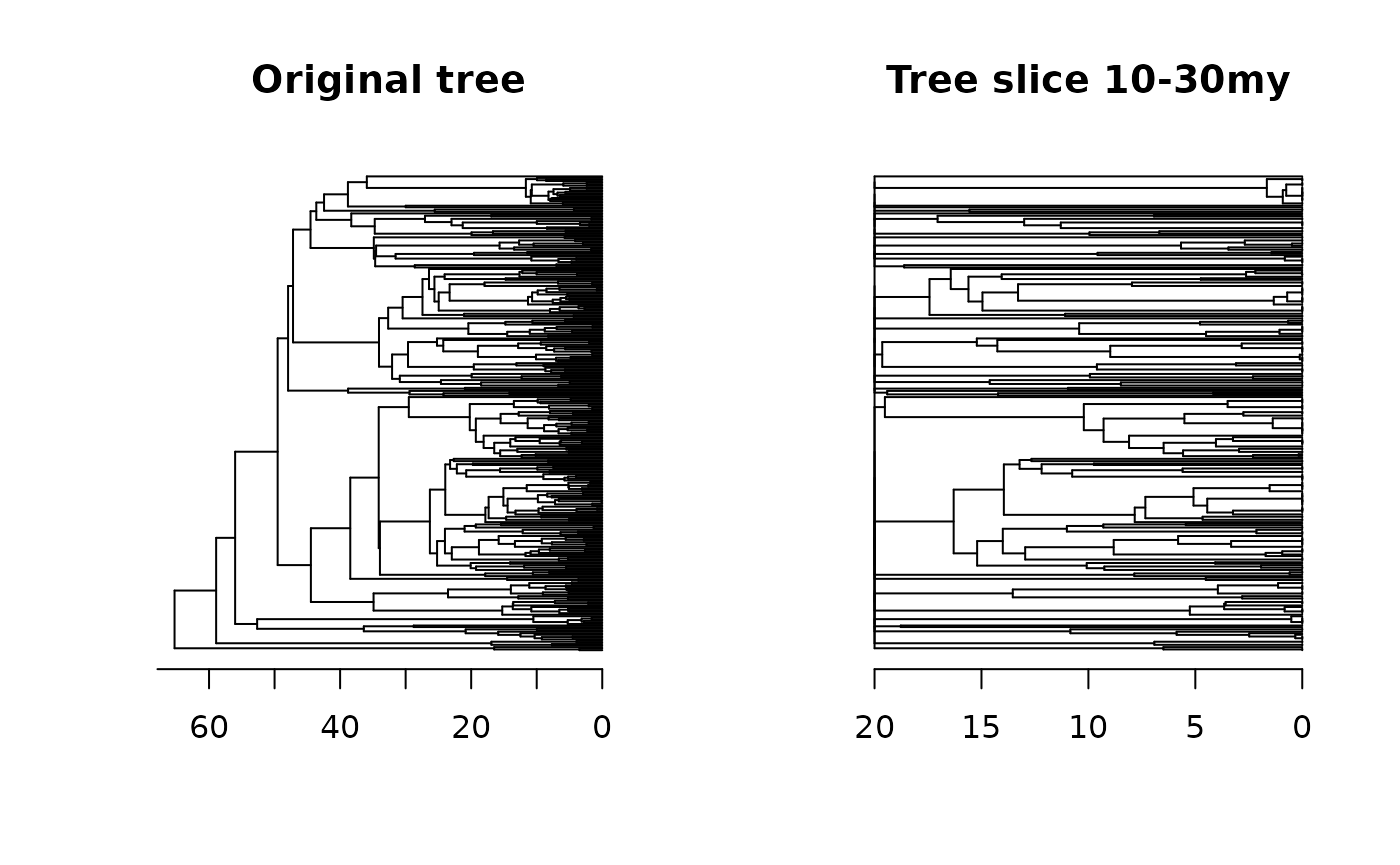
Again, it is essential to note that the absolute depths of the tree will change, but the internal branch lengths will remain the same as the original values (except for the ones that were sliced).
That’s all folks!
References
Jetz, W., Thomas, G. H., Joy, J. B., Hartmann, K. and Mooers, A. O. 2012. The global diversity of birds in space and time. - Nature 491(7424): 444–448. https://doi.org/10.1038/nature11631